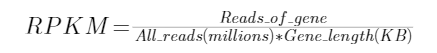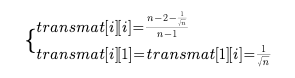注意注意,按照这个方式编译后的R,很多Rcpp开头的包都要源码方式重新编译安装,否则会出错!! 首先,我之所以会做这个事情是因为我某次更新win11之后,发现我的所有R包全都用不了了。于是乎我更新了R
阅读更多...
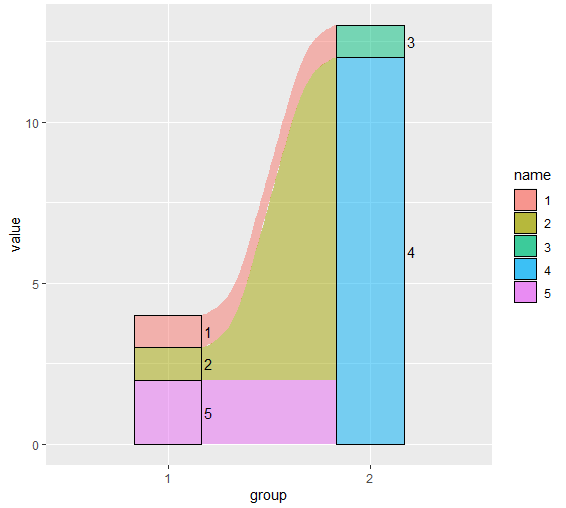
很多时候,我们会有绘制非等高桑基图的需求。 本来我这个文章写了一次了,结果服务器出问题提交失败了。所以没耐心再写一次。 我就直接贴代码说明一下代码吧。 library(ggalluvial) libr
WGCNA,全名加权基因网络分析,目的是更好的使用基因在样本中的表达量去衡量基因与基因之间的关系。 本文不涉及WGCNA的具体实践,也不涉及具体公式推导和证明,有需要者可以直接参考R包WGCNA的教程
在计算RNASeq流程之中的表达量差异,也就是比较不同样本之间的RNA测序reads数的差异 (因为我们认为表达量大的基因,RNA量也会大,反转录的cDNA量也会大,那么测序的结果这部分的cDNA覆盖
quantiles标准化,也就是我将要介绍的最后一个标准化方法,这个标准化方法显然是顾名思义就是根据分位数进行标准化。 该算法在Bioconductor包内的preprocessCore包内,并且是由
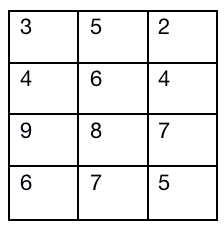
还是和往常一样,首先是要解释一下这个qspline的整体在做一个什么工作。 我们还是要注意这个东西叫做标准化,所以必须是把原数据向某个标准靠拢(映射)。 qspline我没找到网上比较官方的翻译,所以
Bioconductor包之中,有一个标准化方法叫做normalize.AffyBatch.contrasts(),这个方法参照的是一个2001和2003年Magnus Astrand写的两篇文章,其
在affy之中,数据预处理有一个叫做constant normalization的方法。 首先,我要说一个我自己认为的东西。我个人认为这个过程要叫做标准化,而不能叫做归一化。 前一篇文章关于RMA算法
上一篇介绍了RMA算法的背景校正部分。接下来是归一化和最终计算过程。 首先是归一化: 归一化就是将不同组之间的数据统一。因为组之间的数据虽然经过背景校正之后去除了部分噪音。 但是如果某一组自身因为内因
RMA算法是基因芯片数据预处理算法之中一个非常好用而且有效的方法。 本人最近正在学习这个算法,但是发现国内关于这方面的资料是真的很少。 RMA算法分为三个部分, 背景校对 归一化 最终计算表达强度 首
作者邮箱二维码