Bioconductor之affy中qspline normalization(分位点样条插值标准化)方法的介绍
还是和往常一样,首先是要解释一下这个qspline的整体在做一个什么工作。
我们还是要注意这个东西叫做标准化,所以必须是把原数据向某个标准靠拢(映射)。
qspline我没找到网上比较官方的翻译,所以我就斗胆翻译为分位点样条插值标准化了。
根据这个名字就比较好理解在做什么了。
首先是将所有数据,同探针的多组数据根据一定方式计算一个平均值(target),
找到给定参数个分位点,然后根据分位点进行多次的采样,把分位点的探针读数作为x,target作为y进行样条插值,
再将所有数据都通过样条插值函数向平均值映射,从而进行向着平均值标准化。
这个算法实际上是比较简单的,所以完全是用R语言实现的,大家可以去阅读以下。
我接下来按照步骤详细解释这个算法的过程,但实际上没之前的算法内容多,这个是一个比较简单易懂的算法。
首先进行平均数也就是target的计算,在函数里是一个固定的公式

之后进行的其实就是分位点的选取,在调用时可以用p.min和p.max来设置分位点最小和最大位置,是一个小于1大于0的数,以及一个min.offset来控制最小间距。
然后开始按照组循环
每一组里,算法做了一个很棒的事情,因为我们根据样条插值之后,必然有误差,因为样条插值采用的分位点本身可能是有错误的,所以算法里是采样不同的分位点来多次迭代求平均值,用fit.iters参数来控制迭代次数,实际上这个参数在之前选择分位点的时候就用上了,因为每一次迭代都要不同的分位点,所以之前选择分位点的时候就要根据迭代次数选择。
之后每一次迭代拿出一组x和y,根据参数smooth选择是否先对x和y变得光滑,然后便进行样条插值,插值后得到样条插值函数,再将所有的点都带入这个函数得到某个探针输入在插值函数上的值,也就是向target映射,然后除以迭代次数。最后是将所有的迭代结果加起来,因为除以迭代次数了,所以是将所有迭代的结果求了一个平均值。
我个人觉得这个算法是一个比较有效的算法,因为考虑到了分位点自身是有误差的,进行了平均化处理,而且速度很快,整体就是简单的进行多组样条插值而已,而且每一次插值的量也不大,如果大家觉得太慢,可以通过offset和iters来控制一下,减少样条插值点和迭代次数。
这个算法虽然直接看源码因为没注释所以难看懂,但是核心思想不是很复杂,所以我也就介绍到这里了,如果大家有什么问题可以留言。这篇就没之前的写的详细了,调用方式也和之前一样。
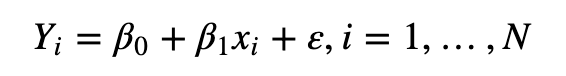



全部评论