RNASeq差异表达中标准化方法集合 (RPKM,FPKM,TPM,DESeq,edgeR)
在计算RNASeq流程之中的表达量差异,也就是比较不同样本之间的RNA测序reads数的差异
(因为我们认为表达量大的基因,RNA量也会大,反转录的cDNA量也会大,那么测序的结果这部分的cDNA覆盖率就会更高,也就是reads数量大)。
但是不同样本之间的数据存在一定整体的偏差,例如某个样本测得深度高,有的测深度低。
不仅仅样本之间,样本内也同样存在问题,例如长度长的基因显然从概率上出现的read数就会多,那么基因与基因之间的差异就不能直接进行数值对比。
为了解决基因与基因之间的偏差,我们引入RPKM,FPKM,TPM三个不同的标准化方法:
RPKM(FPKM), 这两个算法是非常一致的:
首先先介绍一下算法的名字
RPKM
(Reads Per Kilobase of exon model per Million mapped reads, is defined in thisway)
FPKM
(Fragments Per Kilobase of exon model per Million mapped reads, is defined in thisway)
也就是说用了比reads更大范围的fragments
但是我们绝大多数时候遇到的都是reads,所以了解RPKM就可以了,而且他们的计算公式是相同的:
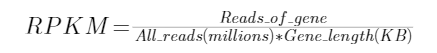
注意我写的公式和百科上或者作者定义的不太一样,因为个人觉得原来的说法有点二义性。
Reads_of_gene:覆盖到该基因位点的reads数量,可能有人会说有一些reads可能同时覆盖到了两个基因,因为可能reads刚好是两个基因的交界,具体该怎么计数呢?这个可以参考HTSeq工具,里面有三种模型任你自由选择。
All_reads:该样本的所有reads数,但是注意这里单位是million是,假如现在我们有1m条reads,那么这里就得用1代入,也就是得除以百万。
Gene_length:该基因的长度,同上单位是KB,也就是除以1000.
其实从公式上,我想大家就能知道这个RPKM是在做什么事情了,因为不同的基因长度不一样,而长度长的基因的reads从概率上就是会更可能出现,所以得除以长度以排除长度对于数量带来的影响。
例如:
| Gene(length) | Number of Reads | RPKM |
| GeneA(16KB) | 11MB | 11MB/(30*16)=22916.67 |
| GeneB(10KB) | 6MB | 20000 |
| GeneC(20KB) | 13MB | 21666.67 |
| ALL reads | 30MB |
2. TPM,该方法是基于上述方法的,是根据RPKM的结果,等比例的平分1MB,直接上例子:
| Gene(length) | Number of Reads | RPKM | TPM |
| GeneA(16KB) | 11MB | 11MB/(30*16)=22916.67 | 1MB*(22916.67/64583.33) =354838.8 |
| GeneB(10KB) | 6MB | 20000 |
309679 |
| GeneC(20KB) | 13MB | 21666.67 | 335483.94 |
| ALL reads | 30MB | 64583.33 |
上述两个方法是处理组内不同基因之间的差异,但是如果我们需要比较的仅仅是组间的差异,那么可以根据下述两个算法进行。
3. 首先介绍简单一点的DESeq:在这里的核心是几何平均数,那么什么是几何平均数呢,请大家参照百度。我这里先说明一下几何平均数的优势:
他相比起算数平均数,对于异常值的影响会更小,因此可以通过几何平均数排除异常值的影响。什么是异常值呢?异常值就是特别大或者特别小的数值。
接下来我根据例子一遍算一遍说算法:
原数据

第一步:对全部数据取log记为Ml

第二步,去除Inf的值
第三步,取出每种样本Ml中的几何平均数

第四步,Ml每种样本减去自己的平均数,记为M2注意这里因为取了对数,
所以本质上是每个样本除以几何平均数后再取对数

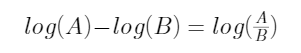
第五步,取出M2每种样本的中位数,再通过指数运算变回原来的数


第六步,原数据每种样本除以第五步各自样本的中位数既完成了DESeq的标准化

4. edgeR标准化方法,这个方法比起EDSeq要更加复杂一点,因为引入了参考样本,而且更加注重考虑异常值的影响。
因为其算法的展示需要数据量比较大才能看到现象,所以我只能通过口述的方式,简单说一下算法的流程。
第一块,选择参考样本
一、去除全是0的基因,并且取log后去除INF值
二、取出所有样本75%分位点数据,记为SF,并取平均记为F。将SF中离F最近的样本作为参考样本。
第二块,计算标准化因子
一、将所有样本同参考样本在每个基因上都做比值,记为P,并且取log,记为LP。对LP进行排序,记为SLP,去除SLP中前后各30%。因为比值大的和小的值说明原数据和参考数据有很大的差异性,这些数据不是用来做标准化的好标准,所以先去掉。
二、将所有样本都同参考样本在每个基因上都求出几何平均数,其实就是每个基因,样本中的值取log,参考样本的值取log,相加除以2。同样排序去除前后5%。这个是因为几何平均数过大和过小的数据说明都是异常值,这也不是好的标准。
三、上述得到的两个数据集取交集,对于交集内的数据去除SLP中每个基因的值,乘以基因的reads数,再除以该样本的总reads数,再通过指数运算得到原来的值,就是标准化因子。
最后所有基因的reads数都除以该标准化因子即可。



全部评论